|
|
| New methods to model global microbial dispersal |
| By Stilianos Louca. February 12, 2021 |
Background - Brownian Motion models of geographic dispersal
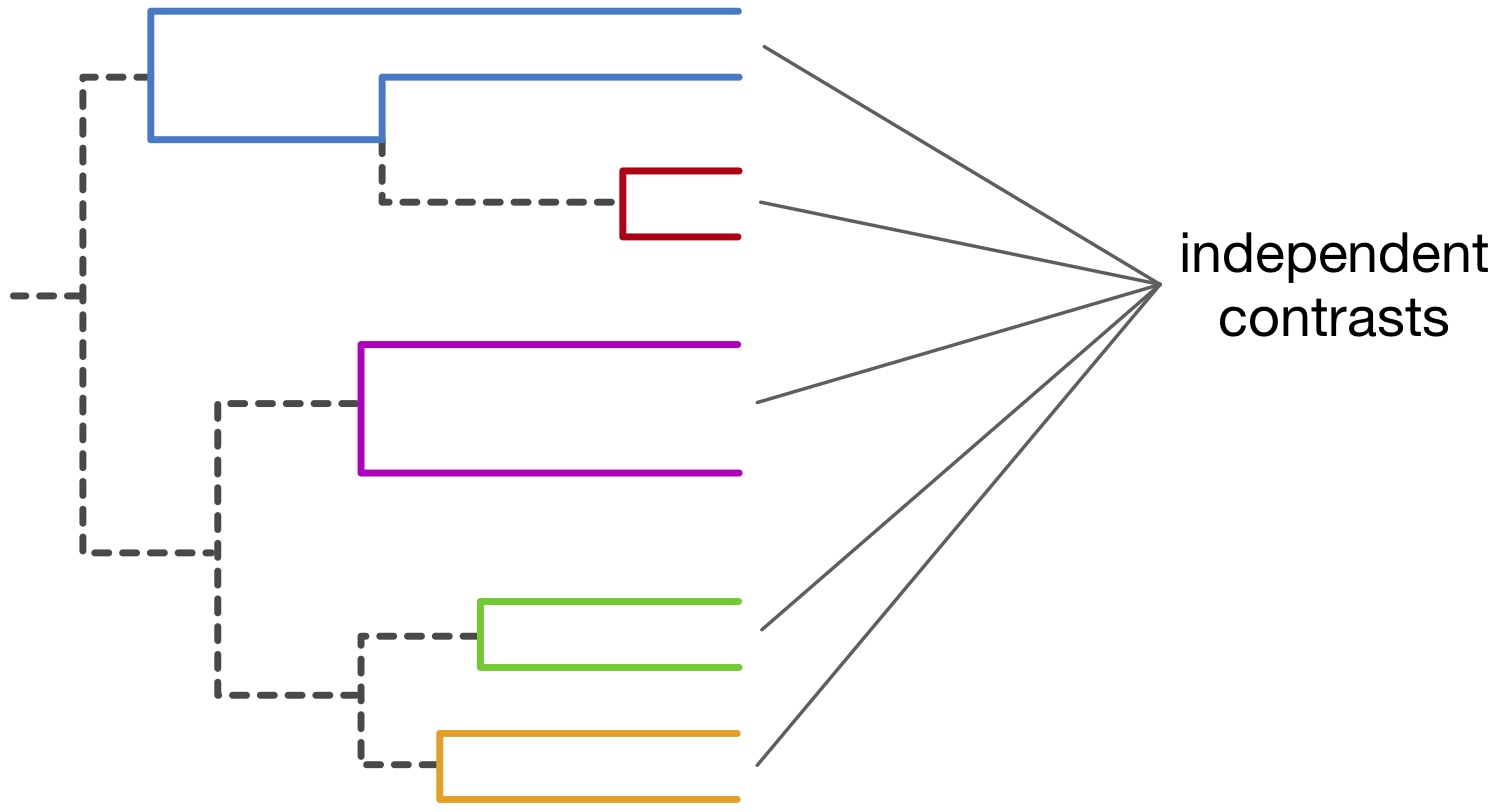
Phylogenetic trees of closely related microorganisms or viruses, i.e. mathematical structures encoding their recent evolutionary relationships, combined with data on their geographic locations, contain information on their geographic dispersal history and dispersal rates. For example, by comparing the geographic distance between related organisms to their evolutionary distance one could in principle estimate the rates at which they disperse over time. Multiple statistical and computational methods have been developed over time to make use of such "phylogeographic" data in order to examine the dispersal of invasive plants and animals, infectious diseases and even cultures. Many of these methods are based on the famous Brownian Motion model for dispersal on a two-dimensional plane, where each point is described by an X and a Y coordinate both of which slowly change over time in random directions. One-, two- or three-dimensional versions of this model have been extremely successful in the natural sciences. In physics, for example, Brownian Motion is commonly used to describe the diffusive motion of gas molecules or ions in solution, and in ecology it is often used to describe random animal movement in space. The main parameter of Brownian Motion is the "diffusivity", which specifies how fast a large number of diffusing points starting at the same location would spread out over time.
Spherical vs. flat space
Classical formulations of Brownian Motion assume a "flat" space, and thus are only suitable for describing geographic dispersal within a small area across which the curvature of Earth can be ignored. If the organisms studied are spread over large areas, such as entire continents or the globe, then Earth's spherical shape cannot be ignored, even if over short time periods dispersal proceeds in Brownian Motion style. For example, a globally dispering virus can spread in one direction and eventually reach its starting point again after having traversed the globe, something that is not possible on a flat surface. In these cases our dispersal models must be modified to account for Earth's spherical geometry. A process that looks like Brownian Motion on the surface of a sphere is called "Spherical Brownian Motion" (SBM).
Until now, no statistical tool existed for estimating SBM diffusivities from a given phylogeny and corresponding coordinates, and no method existed for running simulations of SBM along a given phylogeny. Efficient simulations of dispersal models (i.e., generating hypothetical data in silico based on the model) are needed for evaluating the accuracy of our estimation tools, for obtaining confidence intervals of estimated diffusivities, for statistical hypothesis testing and for determining whether our data deviates substantially from our models.
Improved methods for simulating and fitting Spherical Brownian Motion models
In our latest article, recently published in the journal Systematic Biology, we describe new computational methods for estimating global dispersal rates (diffusivities) using phylogeographic data, based on SBM.
The basic idea behind our estimation methods is to compare the evolutionary and geographic distances between closely related pairs of organisms to estimate how fast they dispersed. Mathematically, this is done using a technique called "maximum likelihood" fitting, whereas we determine the diffusivity that would be most likely to generate the observed combinations of evolutionary and geographic distances across all the pairs of organisms compared. We also present methods for efficiently running simulations of SBM models along any given phylogeny, i.e., generating hypothetical random geographic locations for organisms with specific evolutionary relationships and a specific diffusivity. This possibility of running efficient simulations expands the suite of possible statistical analyses and allows researchers to evaluate the adequacy and accuracy of fitted SBM models.
Examples
Our new methods allow researchers to more rigorously examine the global dispersal of microbes and pathogens, which in turn can aid in disease control and help us understand why some microorganisms are found in certain locations of the world. As an example, we examined hundreds of Cyanobacteria (an important photosynthetic microorganism) sampled from around the world and discovered that Cyanobacteria living in the ocean disperse much faster than their terrestrial relatives, presumably due to rapid ocean circulations. In particular, we estimated that within 1500 years a single marine Cyanobacterial cell lineage is expected to traverse on average about 1500 km, while a terrestrial lineage is expected to traverse on average less than 100 km during that time.
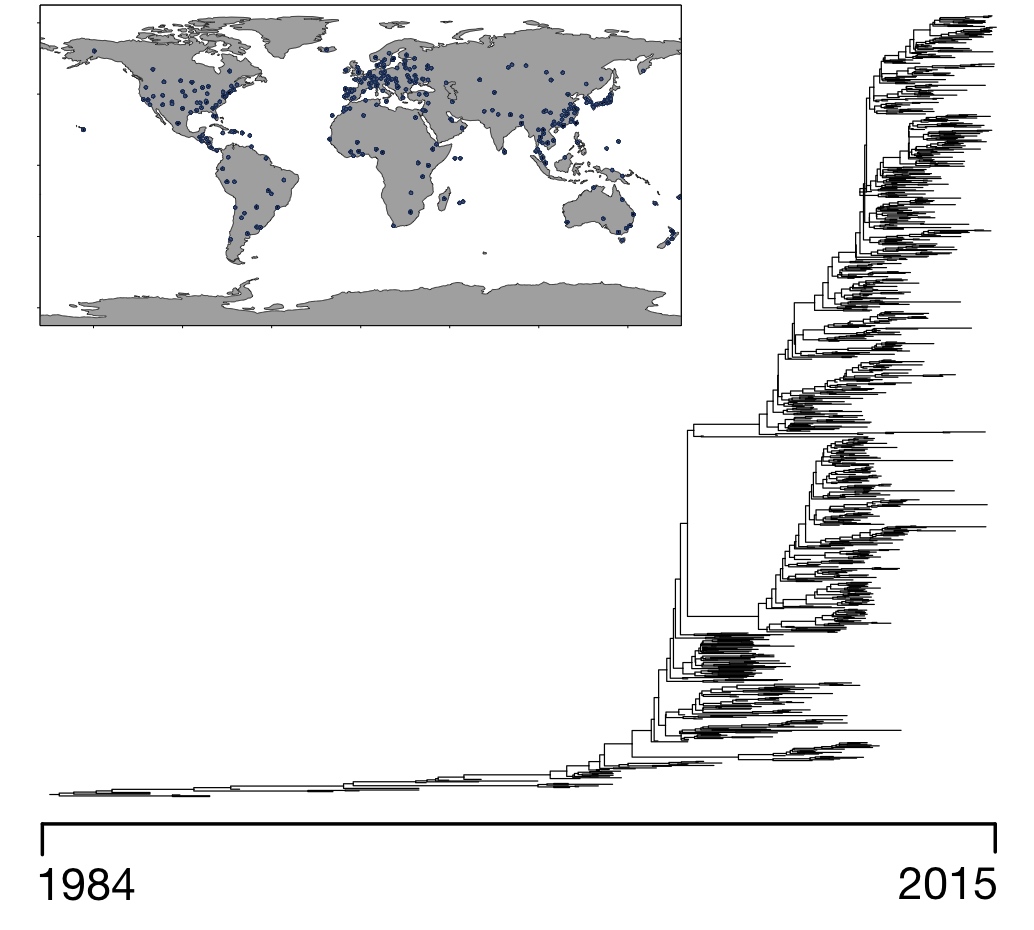
In situations where different organisms are sampled at different time points, for example SARS-CoV-2 strains sampled at various times during the pandemic, we can even reconstruct how the dispersal rates have changed over time. Hence, our methods can be used to check whether specific policies have resulted in a slower or faster geographic spread of a pathogen. For example, when we applied our methods to a phylogeny of 956 Influenza B strains ("Victoria clade") sampled from around the world during the years 1987-2014, we discovered that this clade's geographic dispersal substantially accelerated over time.
Availability
Our new methods are freely available as part of the software package castor.
Full article:
Louca, S. (2021). Phylogeographic estimation and simulation of global diffusive dispersal. Systematic Biology 70:340-359 |
|
 ×
 ×
 ×
|
|
|
Louca lab. Department of Biology, University of Oregon, Eugene, USA
© 2025 Stilianos Louca all rights reserved
|
|
|

