Microorganisms, notably prokaryotes (i.e., bacteria and archaea), are the most ancient, the most widespread and the most ubiquitous form of life on Earth. Their metabolism drives biogeochemical cycles in virtually every ecosystem and has shaped Earth's surface chemistry over billions of years.
Today, prokaryotes are able to utilize a myriad of metabolic pathways to gain energy, thereby occupying extraordinary niches that most other organisms cannot.
Our lab is interested in how prokaryotes impact their environment by catalyzing biogeochemical fluxes and, reciprocally, how ecosystem feedbacks affect microbial diversity.
To answer these questions we use experiments, field surveys, high-throguhput DNA sequencing, bioinformatics, mathematical modeling and statistics.
We also develop novel computational tools, for example for analyzing massive phylogenetic datasets.
Ongoing research topics and some potential graduate theses are summarized below. Future graduate students and postdocs are encouraged to also develop their own research ideas!
Microbial evolution in hot springs
Hot springs are modern analogs of what might have been the earliest type of environments to harbor life on Earth, and are similar to environments likely supporting or having supported life on other planets.
The importance of hot springs to astrobiology and early Earth biology has fueled intense research on the microorganisms inhabiting these environments.
To date, however, little is known about how microorganisms evolve within these environments.
Our lab is conducting molecular sequencing surveys of microbial mats in hot springs across Oregon, Nevada and Yellowstone National Park, to answer fundamental questions about microbial evolution in these extreme and ancient environments.
Due to their isolation, many hot springs also constitute "island-like'' miniature model ecosystems, analogous to the Galapagos, that can serve as experimental replicates to study microbial evolution.
To date, our lab already amassed Terabytes of DNA sequencing data from hundreds of microbial mats, waiting to be analyzed. For some published data see here.
The statistical properties of prokaryotic genomic diversity
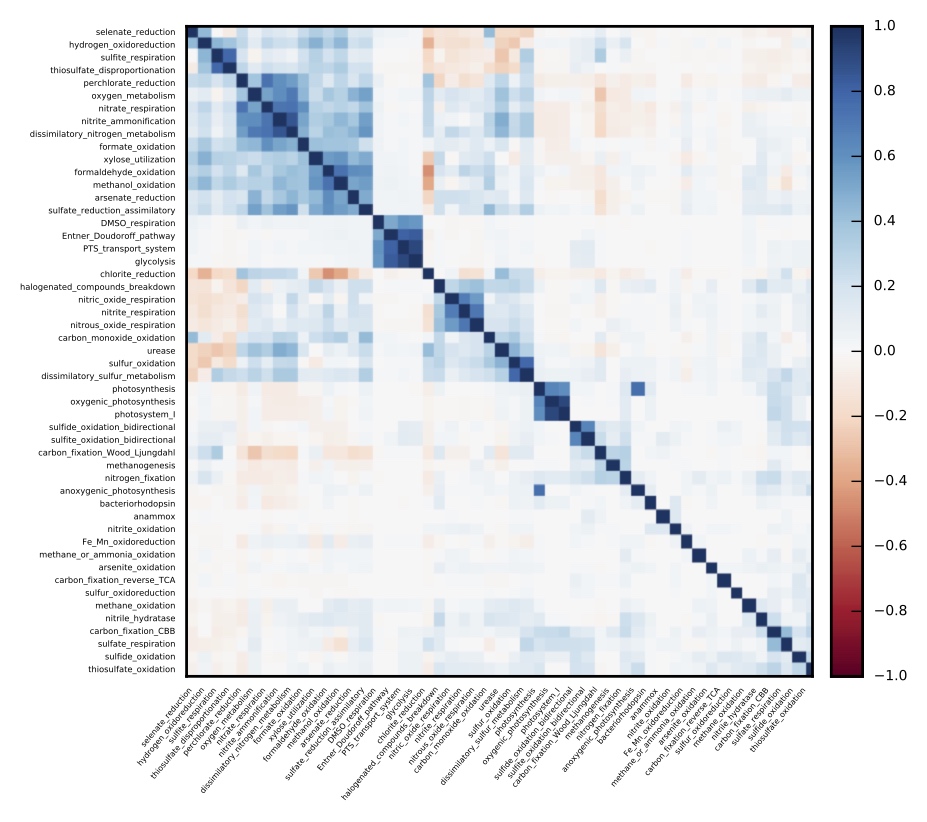
In any given environment, pathways driving redox reactions for energy acquisition can often be shared by numerous coexisting genomes, and sequential pathways are often split across genomes in various alternative combinations. The processes determining pathway distribution patterns across genomes are largely unknown. Can prokaryotic genomic diversity, either within a single ecosystem or at global scales, be described by simple stochastic models of gene content shuffling? Can prokaryotic genomes be described as metacommunities of cooperating but selfishly replicating genes? Do environmental conditions influence the degree to which metabolic pathways are split across coexisting organisms? These questions may be investigated using the myriad of sequenced genomes available in public databases, or using genomes recovered from metagenomes from a single environment.
Development and validation of pathway-centric ecological models
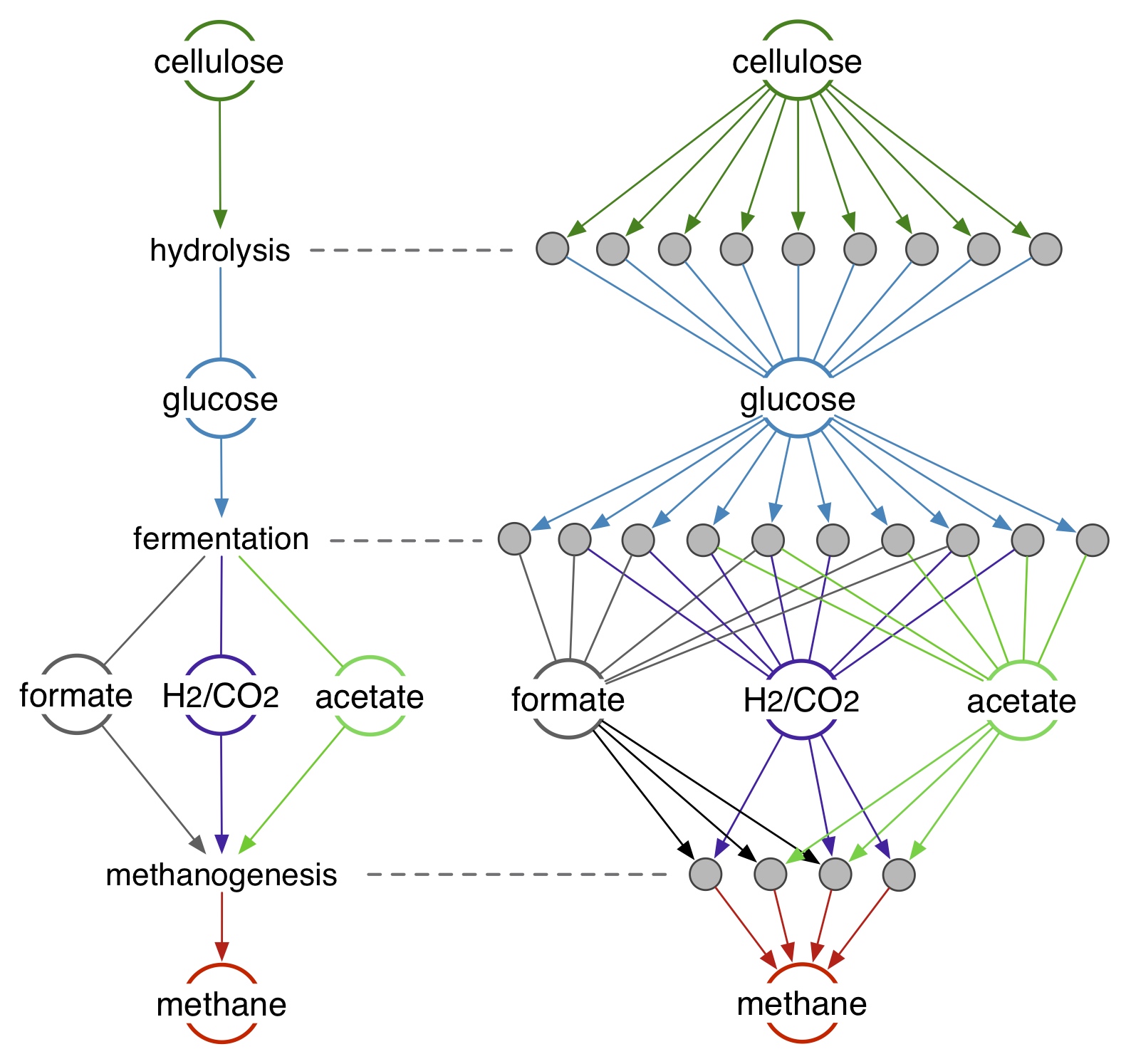
Most metabolic pathways are found in a wide range of microbial taxa, each of which could potentially fill the same metabolic niche. This functional redundancy leads to a partial decoupling between a community's taxonomic composition and bulk metabolic activity. Pathway-centric models, whereby the distribution and activity of pathways are modeled regardless of the species that host them, could yield great insight into microbial metabolic network dynamics at ecosystem scales. Many questions remain along this endeavor. What is the proper format of pathway-centric models, and what are the limits of their applicability? Can pathway-centric models help us understand patterns of DNA, mRNA and protein distributions in nature? Reciprocally, how can metagenomic, metatranscriptomic and metaproteomic data be used to calibrate or validate pathway-centric models? The advent of environmental meta'omic sequencing data makes these questions more relevant than ever.
The role of genomic structure in microbial metabolic networks
What is the role of microbial genomes in microbial metabolic networks at ecosystem scales? How does the cooccurrence and interaction of different genes in genomes influence their metabolic activity and generate deviations from purely gene-centric predictions? These questions can be investigated using mechanistic models, time series monitoring of natural systems as well as microcosm experiments in the lab. Philosophically, this work is central to understanding the role of the various layers at which Life is organized, including individual genes, groups of genes (genomes) and groups of genomes (microbial communities). Practically, this work will help design more accurate biogeochemical models for natural as well as engineered ecosystems.
Experimental and mathematical characterization of microbial system kinetics
Are there universal high-level principles governing the dynamics of microbial metabolic networks, for example in response to nutrient pulses or changing boundary conditions? Microcosm experiments in the lab may help unravel these principles. Metagenomic sequencing and stable isotope probing can be used to characterize the structural and functional responses of microbial systems. System identification techniques from engineering could be used to describe the response of entire microbial systems, just as standard "Monod" response curves are used for single strains. This work would facilitate our understanding of complex microbial system kinetics, beyond just the kinetics of individual strains.
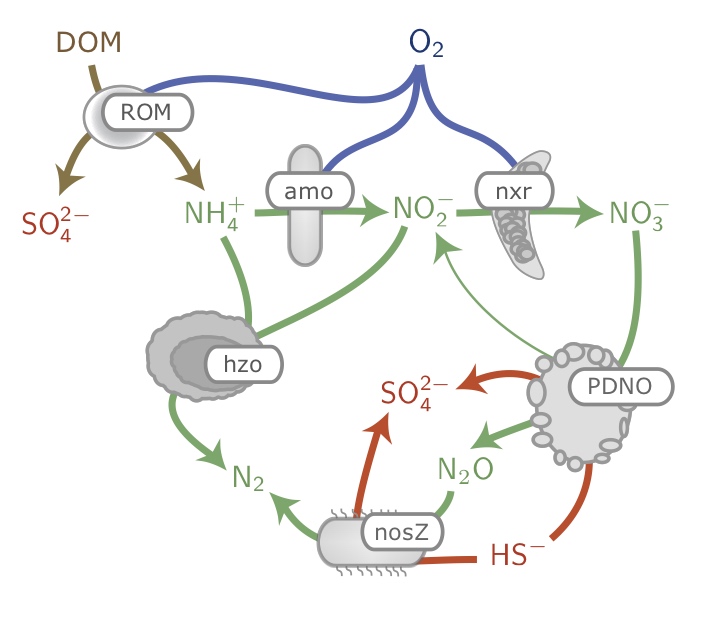
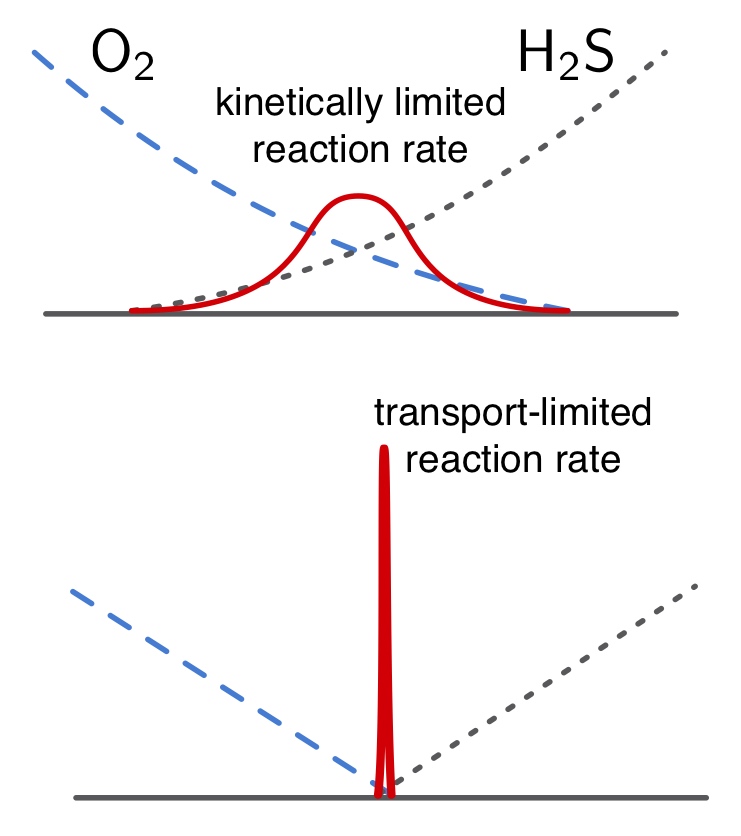
Equilibrium-reaction-transport models for geobiology
The vast majority of geobiological models, such as for nitrogen and sulfur cycling in oxygen minimum zones and sediments, take the form of reaction-transport differential equations in which biologically driven reaction rates are kinetically limited. Such models require extensive kinetic parameterization, which severely limits the applicability of these models to natural systems. Rapid microbial population growth and selection for efficient metabolism, however, can lead to reaction kinetics that are much faster than physical transport processes, thus essentially leading to local thermodynamic equilibrium conditions. Equilibrium-reactive-transport models exploit such local equilibrium conditions to eliminate kinetic parameters, and hold great potential for predicting biogeochemical processes solely based on thermodynamic principles and physics. This project explores the applicability of equilibrium-reactive-transport models to geobiology, including diagenetic processes in sediments and elemental cycling in lakes and the ocean. The project is strongly modeling based, but may also include experimental work.
Development of scalable computational tools
The ongoing explosion of available microbial sequence data presents exceptional opportunities for reconstructing microbial diversification and extinction dynamics over geological time scales, and for unraveling mechanisms that shaped today's extant genomes. These massive datasets, for example including millions of 16S rRNA gene sequences, also present substantial computational challenges, because the majority of existing analytical tools were developed for much smaller datasets. In the Louca lab we develop novel efficient algorithms for handling millions of gene sequences or genomes, for example for constructing and dating trees, detecting horizontal gene transfer events, predicting phenotypes of uncultured organisms and reconstructing diversification dynamics from phylogenies. If you enjoy the intellectual challenge of developing algorithms for computational biology, then this could be the right topic for you!
Charting global microbial diversity
The bulk of microorganisms has never been, and probably will never be, cultured, and culturing is strongly biased towards organisms with specific metabolic traits. Hence our assessment of global microbial diversity is extremely limited and biased. Modern large-scale sequencing surveys can help fill this gap. A goal of ours is to combine existing and future datasets to chart the phenotypic, phylogenetic and geographical distribution of extant microbial diversity, to quantify discovery biases, and to identify relationships between phenotypic and phylogenetic diversity at global scales. This requires the development of new pipelines that can efficiently handle unprecedentedly large datasets. The insight gained from this work will be essential to reconstructing the processes shaping global microbial diversity over geological time scales, for example using Binary State Speciation and Extinction models.
Independent research opportunities for undergraduates
Evaluating model selection techniques in ecology
Information criteria such as AIC and likelihood-ratio-test (LRT) are commonly used in ecology to select among competing models and separate "important" from "unimportant" predictor variables. Despite their widespread use, most of our understanding of the properties and performance of these selection methods are based on linear models and asymptotic considerations (i.e., in the limit of infinitely large datasets). It is not clear, for example, how well the approximate formula commonly used for the statistical significance of an LRT (based on a chi-square distribution) works for finite datasets and complex dynamical models. This project will use simulations of ecological models to systematically investigate the performance of common model selection methods in the presence of finite (realistic) datasets.
Predicting enzyme kinetics using phylogenetic hidden state prediction
Kinetic parameters for the majority of enzymes (e.g., affinity, half-saturation constant, etc) are unknown, but are needed for modeling the metabolism of microorganisms. Physicochemical predictions of kinetics are computationally extremely challenging when only the amino acid sequence is known. Phylogenetic prediction methods, whereby the evolutionary relationships between enzymes are used to estimate an enzyme's properties based on those of close relatives, hold great potential to solving this problem, but remain largely unexplored. This project would use publicly available enzyme sequences and kinetic parameters (e.g., from the BRENDA database) to examine the performance of phylogenetic prediction methods for enzyme kinetics.
Evaluation of tree construction tools at large scales
Massive sequencing datasets covering hundreds of thousands of strains present great opportunities for understanding microbial macroevolution. Unfortunately, existing methods for building phylogenetic trees scale very poorly to such large data sets, and so trade-offs between speed & accuracy become very important. To make things worse, the accuracy of computational tools and the involved trade-offs are poorly understood at these scales. This project's goal is to perform a systematic evaluation of various state-of-the-art alignment/tree-building/tree-dating tools for large datasets common in modern microbiology.
Fundamental trade-offs in enzyme kinetics
It is commonly assumed that fundamental trade-offs exist between various enzyme kinetic parameters, such as affinity and half-saturation constant, however the ubiquity of such trade-offs across enzyme families has never been systematically examined. This project will use enzyme kinetics data across the tree of life from a variety of public databases, to explore and quantify trade-offs between kinetic parameters.
Gene trees vs. species trees
Phylogenetic trees of genes have until recently been used as direct estimates of the underlying species phylogeny, i.e. the evolutionary relationship of the various species from which a gene was sampled. For example, phylogenies constructed using the 16S ribosomal RNA gene are commonly used as a representation of the evolutionary relationships between bacterial species. It is becoming increasingly recognized, however, that gene phylogenies may not fully reflect the underlying speciation history. This project will investigate how much gene trees are expected to differ from the underlying species trees in the case of bacteria, and how much these differences might influence our analyses of bacterial speciation/extinction dynamics over time.
Hypothesis testing in macroevolution
For many taxa the fossil record is too sparse to make meaningful inferences about past speciation and extinction rates, and hence molecular phylogenies of extant-only species are a popular alternative source of information. Recent findings on the identifiability of speciation-extinction models in phylogenetics ("birth-death" models) have called into question the common practice of fitting such models to phylogenies of extant species in order to estimate past speciation and extinction rates. Yet, a debate remains as to whether extant species trees could still be used for testing specific macroevolutionary hypotheses, such as whether speciation has been increasing over time or whether it was influenced by temperature. This project will systematically examine the usefulness of extant species trees for macroevolutionary hypothesis testing.
Phylogenetic relationships between hot spring-associated microbes
Microbial dispersal between hot springs is slower than between most other environments, and hence a geographic restriction of microbial taxa to specific hot springs ("endemism") is relatively likely to encounter. However, the extent and geographic structure of such endemism, especially at global scales, has not yet been systematically examined. This project will build phylogenies for thousands of hot-spring associated bacteria and archaea and investigate the typical taxonomic resolution at which microbes appear endemic to specific hot springs.