
Research overview
Our group uses quantum chemical simulations to study mechanisms of electrical charge conduction, defect formation, and chemical kinetics in hybrid organic-inorganic materials (see Chem. Rev. 2020). We share a particular interest in metal-organic frameworks possessing redox active moieties (see Science 2020), and materials whose properties are not ascertained from crystallography (see J. Am. Chem. Soc. 2021). We also have an ongoing interest in those with unusual physics (e.g. spin liquids, multiferroics; see Nat. Mater. 2020). The group is highly collaborative, working closely with experimentalists both within the USA and beyond.
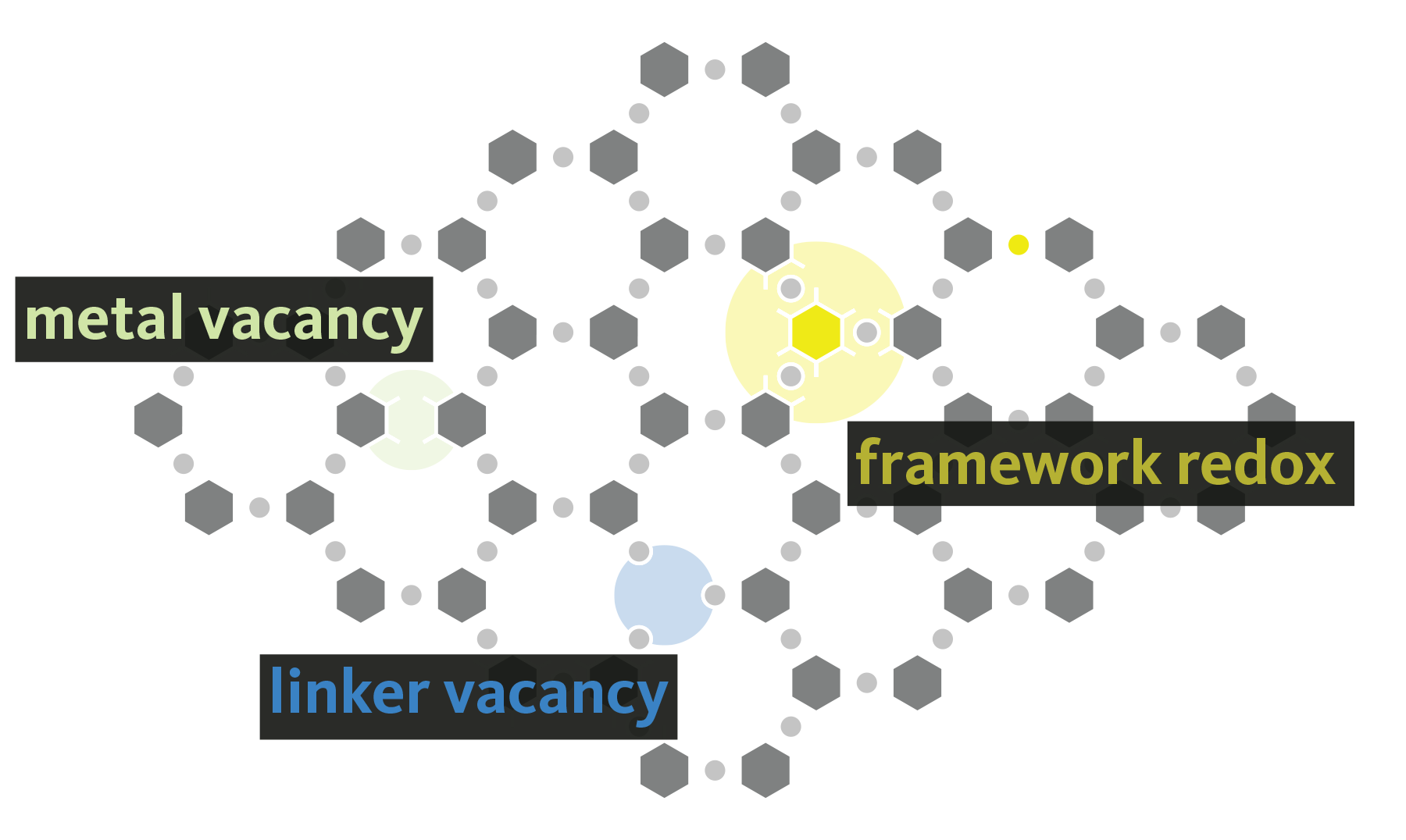
Defects and doping in metal-organic frameworks
Metal-organic frameworks (MOFs) offer a combination of compositional and topological diversity, making them unique platforms for studying structure-function relationships in catalysis and energy conversion. As a result these hybrid materials have garnered increased interest over the past two decades due to their potential applications in gas storage and separation, catalysis, sensors, and as battery materials. But, like most semiconductors, the bulk material derives its properties from defects. These defects come in many forms ranging from local redox events to long range diminished crystallographic order. Substitutions, vacancies, and interstitial defects are considered, with an emphasis on protonic and proton-coupled electronic imperfections. The latter is particularly interesting in both hydrogenation catalysis, and as a mechanicm for tuning the Fermi level in conductive materials. We are also interested in amorphization, dynamic local bonding, and polymorphism in MOFs, but these properties are markedly more rare.
Essentially every member of the group is working towards understand the emergent properties from defects in MOFs. Here are a couple of recent papers on the topic: J. Am. Chem. Soc. 2021 and Chem. Sci. 2021.
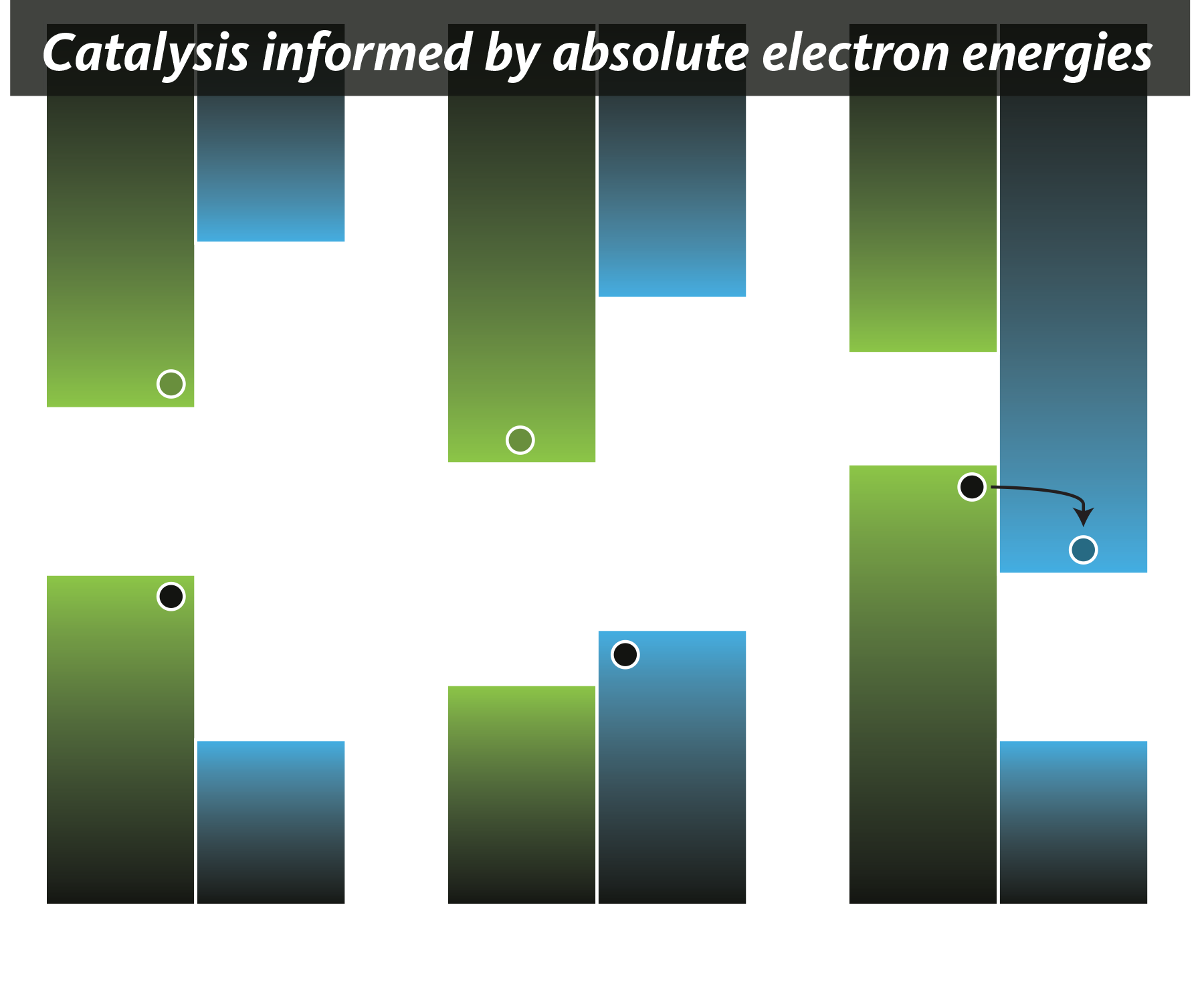
Electron energies and their applications in energy storage and catalysis
The ionization potential/workfunction and electron affinity are two intrinsic properties that play a critical role in determining material reactivity. Predictions of reactivity can be made with knowledge of the alignment of valence/occupied and conduction/unoccupied orbitals. These values are readily obtained from quantum chemical simulations. Three general types of band alignments are possible; Type I where one material's bands sit mid gap relative to a neighbouring material, Type II where charge transfer from one material to another is enabled by the input of some form of energy, and Type III where a material will spontaneously reduce another.
Our group seeks to study the significance of the electronic band gap, the ionization potential, and the electronic affinity in transition metal clusters and cluster-containing materials. In doing so, we seek to develop a general manifold in which to predict material applications in catalysis, as conductors in transistors, and beyond. Here are a couple of recent papers on the topic: Proc. Nat. Acad. Sci. USA 2022 and ACS Materials Lett. 2022.
Modeling and material science of coffee
Since 2014, our group has had a parallel interest in the chemistry and physics of coffee extraction. This topic is suprisingly understudied, despite the coffee sector composing 1.5% of the US gross domestic product in 2015. Our central focus is on the interfacial chemical processes that give rise to differences in chemical compositions in brewed coffee, and developing methods to assess qualities in such beverages. The lab uses a variety of experimental techniques to study these properties, but has expertise in laser diffraction particle size analyses and aqueous electrochemistry. We also use modeling to isolate extraction parameters, allowing for systematic improvement of coffee quality. Naturally, we work closely with industry and are always happy to have visitors in the lab.
Here are a couple of recent papers on the topic: Matter 2020 and A couple more big ones, coming soon!.












